
ライソゾーム病に関して(各論)医師向けGM2ガングリオシドーシス
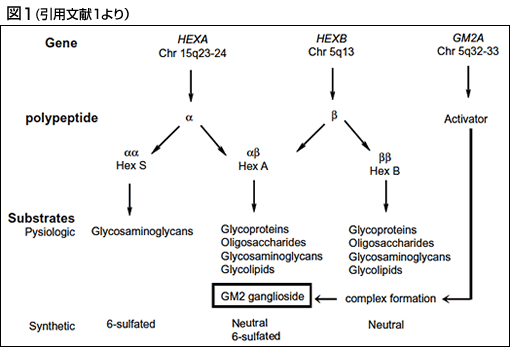
1.概要・欠損酵素(図1参照)
GM2 ガングリオシドーシスは、3つの遺伝子 HEXA, HEXB, GM2A のいずれか一つの遺伝子変異により、GM2 ガングリオシドのライソゾームにおける加水分解が障害され、主として神経細胞のライソゾームに GM2 ガングリオシドが蓄積する疾患である。HEXA 遺伝子(15q23-q24)はβ-ヘキソサミニダーゼ A (β-hexosaminidase A)(Hex A ; EC 3.2.1.52)のαサブユニットを、HEXB遺伝子(5q13)は Hex AH のβサブユニットを、GM2A 遺伝子(5q32-q33)は GM2 活性化蛋白質をそれぞれコードする。 GM2 ガングリオシドのライソゾームにおける加水分解には、酵素である Hex A に加え、基質である GM2 ガングリオシドと複合体を形成する GM2 活性化蛋白質が必要である。ヘキソサミニダーゼには 3 つのアイソザイム-1) αおよびβサブユニットの2量体 (αβ)からなるβ-ヘキソサミニダーゼA (Hex A)、2) βサブユニットの2量体(ββ)からなるβ-ヘキソサミニダーゼB(Hex B)、3) αサブユニットの2量体(αα)からなるヘキソサミニダーゼS(Hex S)があり、それぞれ異なる基質特異性を持つ。
2.遺伝形式
いずれも常染色体劣性遺伝形式である。
3.分類
病因遺伝子により下記の3つの病型に分類される。
テイ-サックス(Tay-Sachs)病(別名:GM2 ガングリオシドーシスB異型)
α-サブユニットをコードする HEXA の遺伝子異常により Hex A 酵素活性が失われることによる。Hex B 酵素活性は正常である。本疾患は1881年に最初に網膜にチェリーレッド斑(Cherry Red Spot)を記載したイギリス人眼科医 Warren Tay と1887年に細胞変化と東ヨーロッパのアシュケナージ系ユダヤ人に高頻度であることを報告したアメリカ人神経科医 Bernard Sachs の名前をとって命名された。
サンドホッフ(Sandhoff)病(別名:GM2 ガングリオシドーシスO異型)
β-サブユニットをコードする HEXB の遺伝子異常の異常により、Hex A と Hex B の酵素活性が同時に失われることによる。
GM2 活性化蛋白質欠損症(別名:GM2 ガングリオシドーシスAB異型)
GM2 活性化蛋白質をコードする GM2A の遺伝子異常により GM2 ガングリオシドと GM2 活性化蛋白質の複合体形成が阻害され、Hex A による GM2 ガングリオシドの分解が障害されることによる。本疾患では Hex A、Hex B ともにその構造、機能に異常はなく、人工基質を用いた Hex A、Hex B の酵素活性測定ではともに正常である。
4.人種差・発生頻度
テイ-サックス(Tay-Sachs)病
| 一般 | (発症頻度) 約360,000新生児に1人。 (ヘテロ保因者) 約280人に1人。 |
|---|---|
| アシュケナージ系ユダヤ人 | (発症頻度) 2,500-3,600新生児に1人。 (ヘテロ保因者) 25-30人に1人。 |
その他にも特定地域や、孤立地域では同様あるいはそれ以上の発生頻度が報告されている。
サンドホッフ(Sandhoff)病
| 非ユダヤ人 | (発症頻度) 約310,000 新生児に1人。 (ヘテロ保因者) 約278人に1人。 |
|---|---|
| ユダヤ人 | (発症頻度) 約1,000,000 新生児に1人。 (ヘテロ保因者) 約500人に1人。 |
その他にも特定地域や、孤立地域では同様あるいはそれ以上の発生頻度が報告されている。
GM2 ガングリオシド活性化蛋白質欠損症
極めて稀。
5.症状
GM2 ガングリオシドは脳の神経組織に多く存在し、細胞膜の主要な構成成分であることから上記の3病型とも、GM2 ガングリオシドは主として神経細胞に蓄積し、患者は進行性の精神運動障害や眼底黄斑部のチェリーレッド斑(cherry-red spot)などの症状を呈する。肝脾腫、骨変化などは認めないことが多い。3病型を臨床症状で区別することは難しい。GM2 ガングリオシドーシスは神経症状の発症時期によりいくつかの病型に分類される。病型の違いは主として遺伝子変異の違いによる残存酵素活性の差による
| 乳児型 | 3-5ヶ月ころまでは正常に発達するが、神経細胞への GM2 ガングリオシドの蓄積にともない、精神運動発達の遅延、退行が認められ始める。眼底黄斑部のチェリーレッド斑が特徴的である。患児は視覚障害、聴覚障害、嚥下困難、痙攣、筋萎縮、痙麻痺をきたす。3歳までには死に至ることが多い。 |
|---|---|
| 若年型 | 稀な病型。2-10歳の小児期に発症する。認知障害、運動障害、言語障害、嚥下障害、運動失調、痙性麻痺をきたし、5-15歳で死にいたることが多い。 |
| 成人型・遅発型 | 稀な病型。20-30歳台前半までに発症する。成人患者ではチェリーレッド斑をしばしば認めず、診断が困難とされる。運動失調、進行性の神経症状が特徴。青年期に構語障害、嚥下障害、運動失調、痙性麻痺、認知障害、精神障害、特に統合失調症様神経症をきたす。患者はしばしば車椅子生活となるが、精神身体障害が支援されれば、寿命を全うしうる。精神症状、痙攣は薬物治療でコントロール可能であることが多い。 |
6.診断
1)酵素活性測定:
血清、白血球、培養細胞(羊水細胞、絨毛細胞を含む)、乾燥ろ紙血を用いて β-ヘキソサミニダーゼ活性測定を行う。
- ■テイ-サックス(Tay-Sachs)病
- Hex A でのみ分解される人工基質 (4-methylumbelliferyl-β-D-N-acetylglucosaminide 6-sulfate [4-MUGS])を用いて Hex A 酵素活性測定を行う。Hex A は熱に不安定、Hex B は熱に安定であることを利用して Hex A、Hex B 両者で分解される人工基質 (4-methylumbelliferyl-β-D-N-acetylglucosaminide [4-MUG])を用いた酵素活性測定も行われる。テイ-サックス病患者の Hex A 活性は欠損、HexB 活性は正常あるいは上昇している。酵素活性測定の結果で異常が認められた場合には、DNA 診断による HEXA 遺伝子変異の検討を行い、偽欠損症の可能性を否定する必要がある。偽欠損症の責任遺伝子変異 (R247W or R249W) を持つ場合では 4-MUGS を用いた酵素活性測定では活性低下を認めるが、天然の GM2 ガングリオシドに対しては正常活性を示し、GM2 ガングリオシドーシス を発症する可能性はない。
- ■サンドホッフ(Sandhoff)病
- 4-MUG を用いて Hex A および Hex B の酵素活性測定が行われる。サンドホッフ病患者では Hex A 活性、Hex B 活性ともに欠損している。
2)DNA 診断
HEXA 遺伝子では105種類、HEXB遺伝子では31種類、GA2A 遺伝子では5種類の遺伝子変異が報告されている。アシュケナージ系ユダヤ人患者の75-80%には1278+TATC, exon 11変異を、15-18%にはIVS12+1G>C, intron 12 変異を認める。 HEXA遺伝子変異では2種類の偽欠損症の責任遺伝子変異アレル739C>T or 745C>T (R247W or R249W) が知られており、これらの変異アレルを持つ場合では人工基質 4-MUGS を用いた酵素活性測定では活性低下を認めるが、天然の GM2 ガングリオシドに対しては正常活性を示す。すなわち HEXA遺伝子に偽欠損症の責任遺伝子変異を持っていても GM2 ガングリオシドーシス を発症する可能性はない。したがって Hex A 酵素活性測定で異常の場合は、DNA 診断による病因変異の検討を行い、偽欠損症の可能性を否定する必要がある。739C>T あるいは745C>T変異は酵素活性測定で保因者と判定されたユダヤ人の2%、非ユダヤ人の35% に認める。また、一般に患者に病因変異を同定できれば、両親の保因者診断が可能になり、出生前診断を含めた遺伝カウンセリングが可能になる。
3)眼科的検査
眼底黄斑部のチェリーレッド斑の同定。乳児型、若年型には必発であるが、成人型・遅発型にはしばしば認めない。
4)病理所見
乳児型の直腸粘膜生検組織の電子顕微鏡所見では、神経節細胞内にスフィンゴ脂質を含む膜成分蓄積の特徴である MCBs (membranous cytoplasmic bodies)を認める。 MCBs は凍結標本で periodic acid-Schiff (PAS) 染色陽性である。
7.治療
現在、GM2 ガングリオシドーシスに対する根本的治療法は確立していない。対症療法、つまり、療育を含め、さまざまな合併症(けいれん、嚥下障害、呼吸障害など)に対する治療が中心となる。下記にあげる治療法に対する基礎研究が進められている。
1)酵素補充療法(Enzyme replacement therapy)
ゴーシェ病1型(非神経型)に対する酵素補充療法の成功をうけて GM2 ガングリオシドーシスに対しても欠損酵素β-ヘキソサミニダーゼAの酵素補充療法が試みられている。しかしながら、GM2 ガングリオシドーシスの病変組織は脳であり、血液脳関門(blood-brain barrier)を超えて酵素を中枢神経系に運ぶことが困難で、十分な治療効果は上がっていない。
2)細胞療法(Cell therapy)
正常なβ-ヘキソサミニダーゼ酵素の供給細胞を移植することによる治療である。その代表的方法として骨髄移植が試みられている。GM2 ガングリオシドーシス患者に対する骨髄移植の報告例はまだわずかではあるが、神経症状発症後1歳時での骨髄移植では酵素補充療法と同様に中枢神経病変に対する治療効果は極めて乏しかったと報告されている。近年、サンドホッフ病モデルマウスに対する神経症状出現前の骨髄移植の治療効果の検討で、脳・脊髄でのβ-ヘキソサミニダーゼ酵素の活性上昇はわずかであったにもかかわらず寿命の延長と神経症状の改善が得られたと報告された。その他の細胞療法として、多分化能を持った神経幹細胞の移植がモデルマウスで試みられている。
3)遺伝子治療(Gene therapy)
欠損遺伝子HEXA, HEXB, GM2A をいかに効率よく神経細胞に導入するかの研究が、さまざまなウイルスベクター、投与経路を用いて行われている。造血幹細胞、神経幹細胞などにレトロウイルス、アデノウイルスを用いて遺伝子を導入後、細胞治療を行う試みもモデルマウスで試みられている。
4)基質抑制療法(Substrate deprivation therapy)
他のライソゾーム病と同様にGM2 ガングリオシドーシスでも蓄積物質の合成を抑制することで治療効果を得ようという試みがなされている。グルコシルセラミド合成酵素の阻害剤である N-butyldeoxynojirimycin (NB-DNJ) をテイ-サックス病モデルマウス、サンドホッフ病モデルマウスに投与し、蓄積物質の減少と寿命の延長が報告されている。この治療法では残存酵素活性の程度が治療効果に影響すると考えられるため、Hex A の残存酵素活性のある遅発型の症例に効果が期待される。N-butyldeoxynojirimycin (NB-DNJ) はヨーロッパでは軽症のゴーシェ病Ⅰ型の治療薬 (Zaversca) として認可されている。
8.疾患モデル動物
| 自然発生モデル | イヌ、ネコ、ブタが報告されている。 |
|---|---|
| 遺伝子改変マウスモデル |
|
9. 引用文献
Gravel, R., Kaback, M.M., Proia, R.L., Sandhoff, K., Suzuki, Ki., and Suzuki, Ku. (2001) The GM2 Gangliosidoses. The Metabolic and Molecular Basis of Inherited Disease. In Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Childs, B. and Vogelstein, B. (eds). McGraw-Hill, New York, pp. 3827-3876.
Aerts JM, Hollak CE, Boot RG, Groener JE, Maas M.
Substrate reduction therapy of glycosphingolipid storage disorders.
J Inherit Metab Dis. 2006;29:449-456.?
Bembi B, Marchetti F, Guerci VI, Ciana G, Addobbati R, Grasso D, Barone R, Cariati R, Fernandez-Guillen L, Butters T, Pittis MG.
Substrate reduction therapy in the infantile form of Tay-Sachs disease.
Neurology. 2006;66:278-280.?
Cachon-Gonzalez MB, Wang SZ, Lynch A, Ziegler R, Cheng SH, Cox TM.
Effective gene therapy in an authentic model of Tay-Sachs-related diseases.
Proc Natl Acad Sci U S A. 2006;103:10373-10378.